Molecular Signatures of Disease
Disease Classification based on high-throughput expression data
With the goal of developing novel methods for disease classification, we have implemented the relative expression classification algorithms, top-scoring pair (TSP) and top-scoring triplet (TST), for the graphics processing unit (GPU). The GPU is a specialized hardware most commonly associated with gaming applications. However, with the NVIDIA’s release of the Compute Unified Device Architecture (CUDA), the GPU became a general-purpose computational device now widely used in computational biology. The GPU architecture emphasizes massive parallelism, in which thousands of parallel threads execute simultaneously. Algorithms that can exploit such parallelism, such as relative expression algorithms, will yield impressive speedups when implemented on GPU hardware. Our implementations of TSP and TST run 200-300X faster than the corresponding CPU implementations of these algorithms, greatly accelerating the pace of discovery.
In addition, we have developed a new general formulation of the relative expression algorithm that is geared towards classification using small numbers of measured features (e.g., microRNAs). This is called the top scoring ‘N’ algorithm (TSN), and it uses a flexible classifier size to expand the permutation and combination space available for classification. TSN relies on an unusual number system known as factoradics to easily translate between permutations of features and decimal numbers. The TSN algorithm has been tested with a number of microarray cancer datasets, demonstrating that the size of the classifier can yield statistically significant differences in cross validation accuracy. In addition, the TSN algorithm has been tested with the Microarray Quality Control II datasets, demonstrating comparable accuracy and very low overfitting when compared to support vector machines, Bayesian methods, logical regression and many other state-of-the-art classification schemes.
DIRAC: Differential Rank Conservation

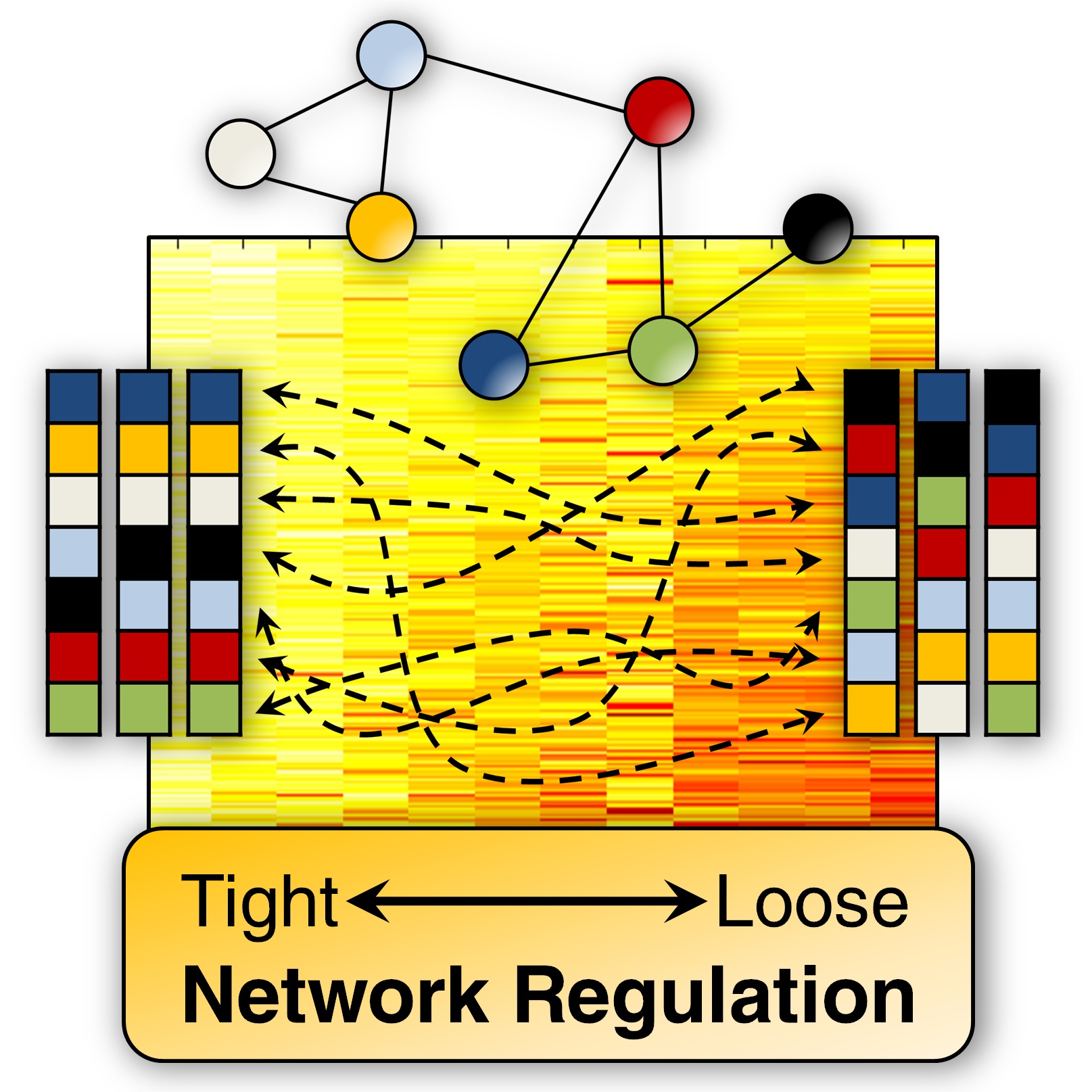
Differential Rank Conservation (DIRAC) is a novel approach for studying gene expression within pathways; this method belongs to a larger family of algorithms designed to identify relative expression (i.e., the ordering among expression values) signatures for diagnosis and prognosis. Specifically, DIRAC provides quantitative measures of how network ordering differs within and between phenotypes. In a pilot study, we examined disease phenotypes including cancer subtypes and neurological disorders, and we identified networks that are tightly regulated, as defined by high conservation of transcript ordering. Interestingly, we observed a strong trend to looser network regulation in more malignant phenotypes and later stages of disease. In addition to the phenotype-level analyses, we performed sample-to-sample analyses and found that, at this level, DIRAC can detect a change in ranking (i.e., shuffling) between phenotypes for any selected network. Finally, we identified variably expressed networks that represent statistically robust differences between disease states, and which serve as signatures for accurate molecular classification.
Building a comprehensive relative expression analysis platform
Public databases such as the NCBI Gene Expression Omnibus contain extensive and exponentially increasing amounts of high-throughput data that can be applied to molecular phenotype characterization. Collectively, these data can be analyzed for such purposes as disease diagnosis or phenotype classification. Relative expression analysis, which includes the Top-Scoring Pair (TSP), Top-Scoring Triple (TST), and Differential Rank Conservation (DIRAC), and Top-Scoring ‘N’ (TSN) approaches, has proven successful in disease classification through analysis of high-throughput gene expression data. These methods have been implemented previously on a variety of computational platforms with varying degrees of usability. To increase the user-base and maximize the utility of these methods, we have developed the program AUREA (Adaptive Unified Relative Expression Analyzer)—a cross-platform tool that has a consistent application programming interface (API), an easy-to-use graphical user interface (GUI), fast running times and automated parameter discovery. AUREA incorporates existing methods, while extending their capabilities and bringing uniformity to their interfaces. We have demonstrated that combining these algorithms and adaptively tuning parameters on the training sets makes these algorithms more consistent in their performance. We have also demonstrated the effectiveness of our adaptive parameter tuner by comparing accuracy across diverse datasets. The unified interface and the adaptive parameter tuning of AUREA provide an effective framework in which to investigate the massive amounts of publically available data by both ‘in silico’ and ‘bench’ scientists.
 hood-price.isbscience.org/research/molecular-signatures-of-disease/
hood-price.isbscience.org/research/molecular-signatures-of-disease/